
For many CROs, diagnostic companies represent an untapped market. When a clinical operations team seeks an outsourcing partner, they need a realistic proposal, fast. Diagnostic companies often find, however, that CROs don’t speak their language. Knowledge gaps about regulatory requirements, the product development process and study design prevail. CRO understanding of the unique needs of diagnostic company studies can narrow those gaps.
Development of diagnostic products depends on complex processes between laboratory scientists and biostatisticians, who comprise the engine that identifies and IP-protects biomarkers, creates and improves assays, and develops and validates algorithms and software. At the end lie reproducible and accurate patient results.
The fuel for this engine is clinical samples. Samples drive every stage of diagnostic development, from biomarker discovery through assay improvement and validation and on to clinical validation and product launch. The number and quality of samples determine timelines and milestones, and the clinical operations team is tasked with hitting those milestones.
Further, budgets for diagnostic development are a fraction of those for device development and orders of magnitude smaller than for drugs. The development process is rapid, requirements may change midway, and clinical operations teams are resource-constrained.
Speaking the diagnostics company’s language is built on understanding of these components:
- The regulatory environment
- The diagnostic product development lifecycle
- Study design
- Site selection
Regulatory Environment
In vitro diagnostics (IVD) which are designed, manufactured and used in a single lab, are regulated by the U.S. Centers for Medicare & Medicaid Services (CMS) through the Clinical Laboratory Improvement Amendments (CLIA). For these laboratory developed tests (LDTs), FDA oversight is optional. IVDs manufactured for distribution to other labs or developed as companion diagnostics in partnership with drug development, however, must undergo premarket FDA review.
The regulatory approach shapes which pre- and post-market studies are needed and, ultimately, the timeline to market. All IVDs must demonstrate analytic validity (i.e., reproducibility and robustness of the assay), but only FDA-regulated products have to show clinical validation (accurate clinical diagnosis as compared to a gold standard) pre-commercialization. Add the increased complexity of IVD quality systems and documentation, and the LDT path to market becomes the speedier and less expensive one.
Product Development Lifecycle
The product development lifecycle for diagnostics is characterized by these phases:
- Biomarker discovery, initial algorithm development, and training
- Testing, ongoing algorithm development, and further training
- Independent test set for internal verification
- Assay and algorithm lock
- Independent test set for clinical validation
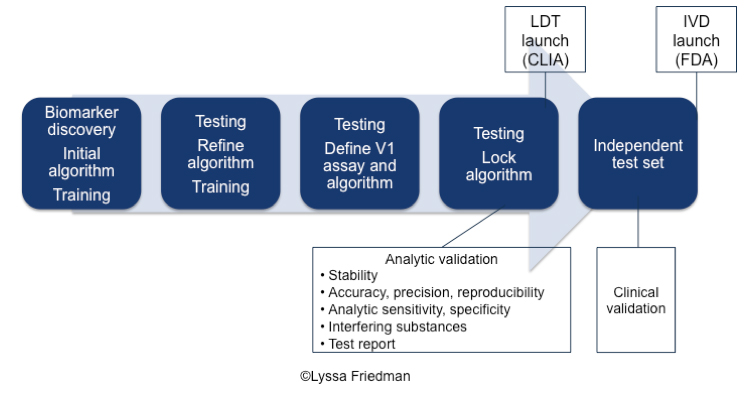
The process is iterative rather than linear. Training, testing, retraining and ongoing testing are part of a continuous feedback loop that relies on rapid cross-functional communication and a constant supply of samples. In fact, delivery of samples can be rate-limiting at any phase.
Study Design
Study goals are simple: accrue quality samples quickly, from the target patient population, which are handled according to stringent protocols and accompanied by verifiable clinical annotation. Studies are regulated by Good Clinical Practices and provide a snapshot of the subject’s clinical condition at the time of sample collection. Because there is no patient intervention, if outcomes are measured it is to support that snapshot, not to evaluate an intervention’s effect.
Because the development system itself needs samples in order to calculate and plan for final algorithmic performance, sample collection studies may lack sample size calculations. As the iterative development process feeds new data about sample and assay stability, the clinical condition, or the gold standard back into the system, modifications to the sample collection protocol may occur. Utilization of multiple sample sources, including retrospective cohorts, is acceptable. Additionally, if the diseased sample population is challenging to accrue, strategies to enrich the sample pool with those that stray from the target commercial population are allowed. Thus, the final statistical analysis plan may be documented internally to encompass samples from all sources rather than embedded in a single sample collection protocol.
If sample collection studies are vehicles for delivering samples, there are a few things they are not:
- Clinical trials. No intervention means no measurement of intervention-related outcomes. No FDA form 1572 is filed, and posting to clinialtrials.gov is not required.
- Long-term. Most sample collection studies employ a single visit for sample collection and retrospective chart review. There are exceptions in which long-term clinical follow-up data is collected or longitudinal samples are collected, if warranted by the product design.
- Obligated to full IRB review. Many sample collection studies, such as those collecting urine, saliva or blood via venipuncture, are considered minimal risk and eligible for expedited IRB review.
- Required to report adverse events or serious adverse events. No patient intervention means no AEs or SAEs.
- Needed to undergo 100% monitoring. Sample collection studies have historically used a risk-based approach to determine which data elements, subjects and sites required monitoring, and in-house data verification can be accomplished using de-identified source data supplied to the clinical operations team.
Site Selection
Because diagnostic products move rapidly to market, study sites are early customers. The clinical operations team is the company’s eyes and ears into intelligence about practice patterns and workflow. Collaboration with key opinion leaders and influencers may weigh more heavily on site selection than study experience or enrollment potential. It is common to handhold strategic sites and investigators, even if underperforming, in order to support strategic goals.
Narrowing the Misunderstanding Gap
When clinical operations teams need outsourcing, they want a partner who not only understands the diagnostics environment but can also speak the diagnostics language. To craft a proposal that shows diagnostic literacy, consider:
- Manage the sample. Demonstrate know-how about sample collection, handling and chain-of-custody documentation.
- Foster strategic relationships. Relationships drive success. Demonstrate the ability to support priority sites even when they underperform. Communicate transparently. Understand that the sponsor owns the relationships.
- Offer appropriate services. Lab work and biostatistics are the creative force behind the product and intellectual property and few companies outsource those services. Site qualification and training is streamlined, and minimal monitoring is needed. If a service isn’t requested, don’t add it to the proposal.
- Move quickly. Activate study sites that have the patient population to achieve goals. If goals slip, take responsibility and queue up solutions.
- Stay flexible. Assume that protocols, sample handling instructions and case report forms will change. Edit documents and implement changes fast.
- Be budget-conscious. Acknowledge resource limits and provide budget transparency at the line-item level. If the budget is below the vendor’s threshold, save time, be honest and suggest a referral. For some CROs, diagnostic company clients do not make business sense.
Diagnostics companies need CRO partners with diagnostics literacy. Speaking the diagnostics language is a CRO’s first step in winning their business.